publications
2024
- Recurrent evolution and selection shape structural diversity at the amylase locusDavide Bolognini, and 9 more authorsNature, Sep 2024Publisher: Nature Publishing Group
The adoption of agriculture triggered a rapid shift towards starch-rich diets in human populations1. Amylase genes facilitate starch digestion, and increased amylase copy number has been observed in some modern human populations with high-starch intake2, although evidence of recent selection is lacking3,4. Here, using 94 long-read haplotype-resolved assemblies and short-read data from approximately 5,600 contemporary and ancient humans, we resolve the diversity and evolutionary history of structural variation at the amylase locus. We find that amylase genes have higher copy numbers in agricultural populations than in fishing, hunting and pastoral populations. We identify 28 distinct amylase structural architectures and demonstrate that nearly identical structures have arisen recurrently on different haplotype backgrounds throughout recent human history. AMY1 and AMY2A genes each underwent multiple duplication/deletion events with mutation rates up to more than 10,000-fold the single-nucleotide polymorphism mutation rate, whereas AMY2B gene duplications share a single origin. Using a pangenome-based approach, we infer structural haplotypes across thousands of humans identifying extensively duplicated haplotypes at higher frequency in modern agricultural populations. Leveraging 533 ancient human genomes, we find that duplication-containing haplotypes (with more gene copies than the ancestral haplotype) have rapidly increased in frequency over the past 12,000 years in West Eurasians, suggestive of positive selection. Together, our study highlights the potential effects of the agricultural revolution on human genomes and the importance of structural variation in human adaptation.
- Structural variation in humans and our primate kin in the era of telomere-to-telomere genomes and pangenomicsJoana L. Rocha*, and 2 more authorsCurrent Opinion in Genetics & Development, Aug 2024
Structural variants (SVs) account for the majority of base pair differences both within and between primate species. However, our understanding of inter- and intra-species SV has been historically hampered by the quality of draft primate genomes and the absence of genome resources for key taxa. Recently, advances in long-read sequencing and genome assembly have begun to radically reshape our understanding of SVs. Two landmark achievements include the publication of a human telomere-to-telomere (T2T) genome as well as the development of the first human pangenome reference. In this review, we first look back to the major works laying the foundation for these projects. We then examine the ways in which T2T genome assemblies and pangenomes are transforming our understanding of and approach to primate SV. Finally, we discuss what the future of primate SV research may look like in the era of T2T genomes and pangenomics.
- The complete sequence and comparative analysis of ape sex chromosomesKateryna D. Makova, and 83 more authorsNature, Jun 2024
Reference assemblies of great ape sex chromosomes show that Y chromosomes are more variable in size and sequence than X chromosomes and provide a resource for studies on human evolution and conservation genetics of non-human apes.
- Complete sequencing of ape genomesDongAhn Yoo, and 122 more authorsJul 2024Pages: 2024.07.31.605654 Section: New Results
We present haplotype-resolved reference genomes and comparative analyses of six ape species, namely: chimpanzee, bonobo, gorilla, Bornean orangutan, Sumatran orangutan, and siamang. We achieve chromosome-level contiguity with unparalleled sequence accuracy (\textless1 error in 500,000 base pairs), completely sequencing 215 gapless chromosomes telomere-to-telomere. We resolve challenging regions, such as the major histocompatibility complex and immunoglobulin loci, providing more in-depth evolutionary insights. Comparative analyses, including human, allow us to investigate the evolution and diversity of regions previously uncharacterized or incompletely studied without bias from mapping to the human reference. This includes newly minted gene families within lineage-specific segmental duplications, centromeric DNA, acrocentric chromosomes, and subterminal heterochromatin. This resource should serve as a definitive baseline for all future evolutionary studies of humans and our closest living ape relatives.
- TRACKing Tandem Repeats: a customizable pipeline for identification and cross-species comparisonsCarolina L. Adam, and 3 more authorsSep 2024Pages: 2024.09.27.615531 Section: New Results
Summary TRACK is a user-friendly command-line pipeline designed to consolidate the discovery and comparison of tandem repeats (TRs) across species. TRACK facilitates the cataloging and filtering of TRs based on reference genomes or T2T transcripts, and applies reciprocal LiftOver and sequence alignment methods to identify putative homologous TRs between species. For further streamlined analyses, TRACK can be used to genotype TRs and subsequently estimate and plot basic population genetic statistics. By integrating existing tools into one integrated workflow, TRACK enhances TR analysis accessibility and reproducibility, while offering flexibility for the user. Availability The TRACK toolkit with step-by-step tutorial is freely available at https://github.com/caroladam/track.
2023
-
 North African fox genomes show signatures of repeated introgression and adaptation to life in desertsJoana L. Rocha, and 10 more authorsNature Ecology & Evolution, Jun 2023
North African fox genomes show signatures of repeated introgression and adaptation to life in desertsJoana L. Rocha, and 10 more authorsNature Ecology & Evolution, Jun 2023Elucidating the evolutionary process of animal adaptation to deserts is key to understanding adaptive responses to climate change. Here we generated 82 individual whole genomes of four fox species (genus Vulpes) inhabiting the Sahara Desert at different evolutionary times. We show that adaptation of new colonizing species to a hot arid environment has probably been facilitated by introgression and trans-species polymorphisms shared with older desert resident species, including a putatively adaptive 25 Mb genomic region. Scans for signatures of selection implicated genes affecting temperature perception, non-renal water loss and heat production in the recent adaptation of North African red foxes (Vulpes vulpes), after divergence from Eurasian populations approximately 78 thousand years ago. In the extreme desert specialists, Rueppell’s fox (V. rueppellii) and fennec (V. zerda), we identified repeated signatures of selection in genes affecting renal water homeostasis supported by gene expression and physiological differences. Our study provides insights into the mechanisms and genetic underpinnings of a natural experiment of repeated adaptation to extreme conditions.
- Genetic basis of aposematic coloration in a mimetic radiation of poison frogsTyler Linderoth, and 12 more authorsApr 2023
The evolution of mimicry in a single species or population has rippling inter and intraspecific effects across ecological communities, providing a fascinating mechanism of phenotypic diversification. In this study we present the first identification of genes underlying Müllerian mimicry in a vertebrate, the Peruvian mimic poison frog, Ranitomeya imitator. We sequenced 124 R. imitator exomes and discovered loci with both strong divergence between different mimetic morphs and phenotypic associations within an intraspecific admixture zone, implicating mc1r, asip, bsn, retsat, and krt8.2 in the evolution of mimetic color phenotypes. We confirmed these associations for most candidate genes through linkage mapping in a lab-reared pedigree. We also sequenced transcriptomes from the model species, allowing tests for introgression and revealing that the mimetic resemblance between R. imitator and the models evolved independently. Selection analyses of the candidate genes show that the mimicry phenotypes likely have evolved through selective sweeps acting on polygenic variation. Our results suggest that the evolutionary origins and molecular mechanisms underlying mimicry phenotypes in vertebrates may be radically different from those previously documented in invertebrates such as the iconic Heliconius butterfly mimicry complex. One Sentence Summary Müllerian mimicry evolved through independent selective sweeps on color and pattern loci in the mimic poison frog.

2022
-
 African climate and geomorphology drive evolution and ghost introgression in sable antelopeJoana L. Rocha, and 7 more authorsMolecular Ecology, Apr 2022
African climate and geomorphology drive evolution and ghost introgression in sable antelopeJoana L. Rocha, and 7 more authorsMolecular Ecology, Apr 2022The evolutionary history of African ungulates has been explained largely in the light of Pleistocene climatic oscillations and the way these influenced the distribution of vegetation types, leading to range expansions and/or isolation in refugia. In contrast, comparatively fewer studies have addressed the continent’s environmental heterogeneity and the role played by its geomorphological barriers. In this study, we performed a range-wide analysis of complete mitogenomes of sable antelope (Hippotragus niger) to explore how these different factors may have contributed as drivers of evolution in southcentral Africa. Our results supported two sympatric and deeply divergent mitochondrial lineages in west Tanzanian sables, which can be explained as the result of introgressive hybridization of a mitochondrial ghost lineage from an archaic, as-yet-undefined, congener. Phylogeographical subdivisions into three main lineages suggest that sable diversification may not have been driven solely by climatic events affecting populations differently across a continental scale. Often in interplay with climate, geomorphological features have also clearly shaped the species’ patterns of vicariance, where the East Africa Rift System and the Eastern Arc Mountains acted as geological barriers. Subsequent splits among southern populations may be linked to rearrangements in the Zambezi system, possibly framing the most recent time when the river attained its current drainage profile. This work underlines how the use of comprehensive mitogenomic data sets on a model species with a wide geographical distribution can contribute to a much-enhanced understanding of environmental, geomorphological and evolutionary patterns in Africa throughout the Quaternary.

2021
-
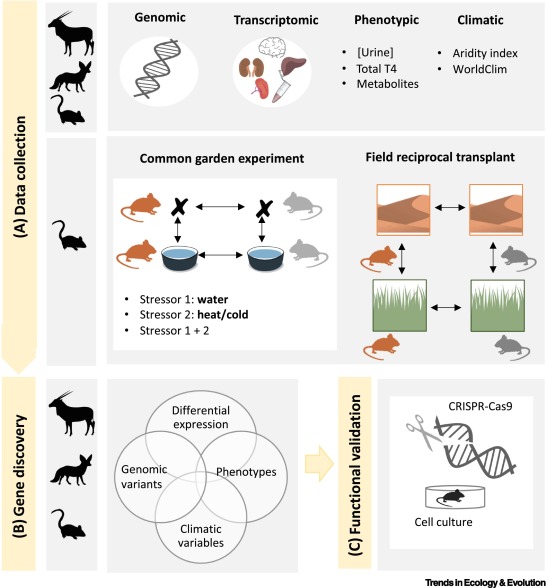 Life in Deserts: The Genetic Basis of Mammalian Desert AdaptationJoana L. Rocha, and 3 more authorsTrends in Ecology & Evolution, Apr 2021
Life in Deserts: The Genetic Basis of Mammalian Desert AdaptationJoana L. Rocha, and 3 more authorsTrends in Ecology & Evolution, Apr 2021Deserts are among the harshest environments on Earth. The multiple ages of different deserts and their global distribution provide a unique opportunity to study repeated adaptation at different timescales. Here, we summarize recent genomic research on the genetic mechanisms underlying desert adaptations in mammals. Several studies on different desert mammals show large overlap in functional classes of genes and pathways, consistent with the complexity and variety of phenotypes associated with desert adaptation to water and food scarcity and extreme temperatures. However, studies of desert adaptation are also challenged by a lack of accurate genotype-phenotype-environment maps. We encourage development of systems that facilitate functional analyses, but also acknowledge the need for more studies on a wider variety of desert mammals.
- Convergent evolution of increased urine‐concentrating ability in desert mammalsJoana L. Rocha, and 3 more authorsMammal Review, Mar 2021
1. One of the most celebrated textbook examples of physiological adaptations to desert environments is the unique ability that desert mammals have to produce hyperosmotic urine. Commonly perceived as an adaptation mainly observed in small rodents, the extent to which urine-concentrating ability has evolved independently in distinct mammalian lineages has not previously been assessed using modern phylogenetic approaches. 2. We review urine-concentrating ability data from the literature in 121 mammalian species with geographic ranges encompassing varying climatic conditions. We explicitly test the general hypothesis that desert-dwelling mammals have evolved greater ability to concentrate urine than non-desert species, controlling for body mass, phylogenetic affinity and other covariates. 3. Ancestral state reconstruction across our dataset’s phylogeny shows that the ability to produce hyperosmotic urine, measured as maximum urine osmolal-ity, has evolved convergently in mammalian species with geographic ranges characterised by low mean annual aridity index. 4. Phylogenetic generalised least-squares (PGLS) models show that the mean annual aridity index of a species’ geographic range largely predicts its urine-concentrating ability, even when accounting for body mass differences, phylogenetic correlations , the specific condition under which urine osmolality was measured, the method used to measure urine osmolality, and the species’ diet. 5. In contrast, we find much weaker correlations between mass-adjusted basal metabolic rate and environmental variables when analysing 84 of the species included in the urine osmolality analysis. 6. Taken together, our results not only show that desert mammals effectively concentrate more urine than non-desert mammals, but further suggest that aridity is likely to have been one of the main selective pressures leading to increasing maximum urine-concentrating ability and driving its repeated evolution in different desert mammalian lineages.
